
Hirschsprung Disease : Pathogenesis



Comments:
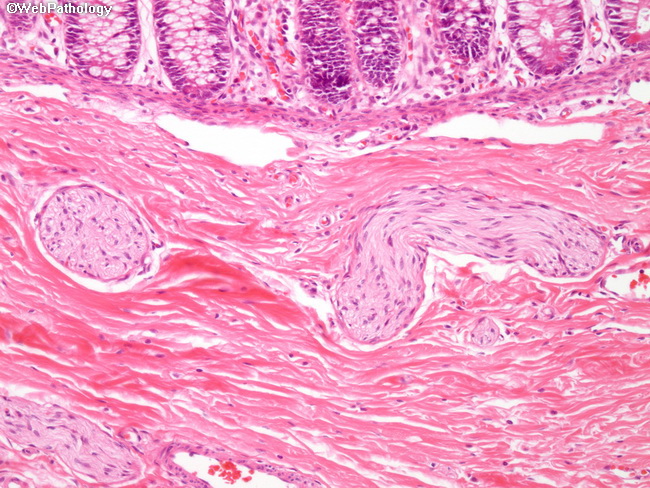
Pathogenesis of Hirschsprung Disease: The exact cause of defective migration of neural crest cells is unknown; however, a genetic component plays a major role. Most of the familial cases and 15% of sporadic cases have heterozygous loss-of-function mutations in RET proto-oncogene. Its protein product assists neural crest cells in their migration through the digestive tract during embryogenesis. Mutations in several other genes that play a role in the normal development of enteric neuronal plexus have been identified. They include EDNRB (encodes endothelin receptor type B), EDN3 (encodes endothelin-3), and GDN3 (glial cell-derived neurotrophic factor). These genes accounts for only 30-40% of cases. Other modifying genes and environmental factors may also have a role. Terminology: Hirschsprung is subdivided into 4 types based on the length and anatomic distribution of the aganglionic segment. Rectum is almost always involved. The extent of involvement ranges from rectum and sigmoid to almost the entire colon. 1) Short segment: Involvement of rectum and distal sigmoid; about 75% to 80% of cases. 2) Long segment : Involvement extends upto hepatic flexure; about 15% of cases. 3) Total colonic aganglionosis (Zuezler-Wilson syndrome): Aganglionosis extends all the way to the cecum with some cases involving even the distal small bowel; 5% of cases. 4) Ultrashort segment: Involves only the distal rectum near the anal sphincter. It is a controversial entity. The image is a higher magnification of the previous image showing hypertrophic submucosal nerve fibers and the absence of ganglion cells in submucosal (Meissner) plexus.