
Rosai-Dorfman Disease : Clinical Features



Comments:
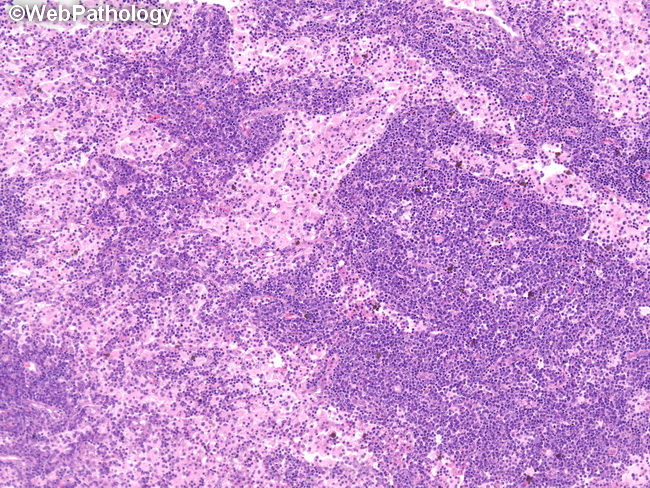
Rosai-Dorfman disease (RDD) usually presents in children and young adults. The median age at diagnosis is 20 years. There is a slight male preponderance and African-Americans are more commonly affected. It has a predilection for lymph nodes in the head and neck region. It usually presents with massive, bilateral, painless enlargement of cervical lymph nodes. Other lymph node sites can also be involved. Hepatosplenomegaly is rare. There is fever, night sweats, weight loss, leukocytosis, elevated erythrocyte sedimentation rate, mild normochromic normocytic anemia, and polyclonal hyperglobulinemia. The clinical presentation may mimic a lymphoma. Unlike Langerhans cell histiocytosis, there are no osteolytic bone lesions but sclerotic bone lesions may sometimes occur. Extranodal sites are involved in 25% to 40% of cases. The disease has been reported in virtually every body site/tissue. The most common extranodal sites include skin, central nervous system, upper respiratory tract, orbit, bones, breast, salivary glands, and gastrointestinal tract. Central nervous system involvement in RDD presents as dura-based, extra-axial lesions of the cranium. Spinal cord or intracerebral involvement is uncommon. The neurological symptoms depend upon the location of the lesion and most commonly include seizures and headaches. Cutaneous Rosai-Dorfman disease (CRDD) makes up about 9% of cases of RDD. It is clinically distinct from RDD and it may be a different but related condition. The patients are older (usually in their 4th decade) as compared to RDD and there is a female predilection. CRDD is more common in Caucasians and Asians. There is usually no adenopathy. The image shows effacement of nodal architecture by expanded sinusoids containing abundant pale-staining histiocytes.