
PanNET : Introduction


Comments:
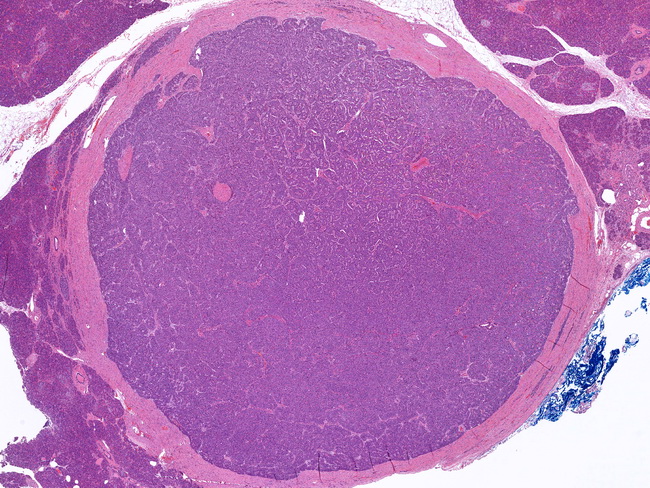
INTRODUCTION: Pancreatic neuroendocrine tumors (PanNETs) are low-grade well-differentiated malignant tumors with potential for local invasion and/or metastases. They have previously been known as Islet Cell Tumors. They measure ≥ 0.5 cm in size; PanNETs smaller than 0.5 cm are referred to as pancreatic neuroendocrine microadenomas. PanNETs can be subdivided into functioning and non-functioning tumors. Non-functioning PanNETs make up 70-80% of all PanNETs. By definition, they are not associated with a clinical hormonal syndrome. They may be discovered incidentally or become apparent due to large size, invasion of adjacent organs, or metastasis. Pancreatic neuroendocrine microadenomas are usually non-functioning. The term "non-functioning" is a misnomer, because even though there are no hormonal manifestations, most tumors synthesize one or more peptide hormones or biogenic substances at subclinical level (e.g. PP, chromogranin A, somatostatin) which can be demonstrated immunohistochemically. Functioning PanNETs are associated with a clinical syndrome caused by abnormal hormonal secretion by the tumor cells. The most common are insulinomas and gastrinomas; others include Glucagonoma, VIPoma; tumors secreting ACTH, GHRH, PTHrP, and CCK. Many tumors produce multiple hormones; but the clinical syndrome is attributable to only one hormone. A functioning PanNET is defined by clinical presentation and not just on the basis of immunohischemistry alone. The specific hormone/s produced by the tumor are of no clinical or prognostic significance; therefore, immunohistochemical staining for peptide hormones is only of academic interest and is not required for diagnosis. About this image: Low magnification scan showing a small, well-circumscribed PanNET surrounded by normal pancreatic parenchyma.