Apr 2020
Insulinoma
Reviewer(s): Dharam Ramnani, MD
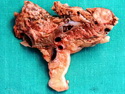
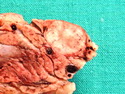
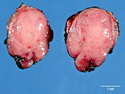
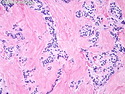
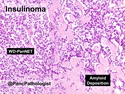
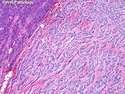
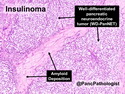
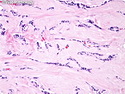
Insulinoma is the most common functioning PanNET. Most are sporadic; 10% are seen in MEN 1 or familial insulinomatosis (mutations in MAFA gene). About 2% are extrapancreatic. Peak incidence is in the 6th decade. Uncontrolled insulin secretion causes severe hypoglycemia with autonomic symptoms (palpitations, tremor, sweating, and hunger) and neuroglycopenic symptoms (weakness, confusion, agitation, seizures, loss of consciousness). Whipple triad (hypoglycemic symptoms, fasting plasma glucose <50 mg/dl, and symptom relief with glucose) is not specific for insulinoma. Laboratory findings: fasting plasma glucose <50 mg/dl; elevated serum insulin, C-peptide, and proinsulin. CT, MRI, endoscopic ultrasound, Octreotide scintigraphy, and PET/CT with radiolabeled GLP-1 receptor analog show well-defined hypervascular tumor. Sporadic tumors are solitary, usually <2.0 cm, and well-demarcated. Tumors that metastasize (10%) tend to be larger (mean diameter 5-6 cm). Patients with MEN 1 or familial insulinomatosis have multiple small tumors. Tumor cells are monomorphic with "salt and pepper" chromatin and arranged in trabecular, tubulo-acinar and solid growth patterns. Stroma is often hyalinized and may show amyloid deposits (5% of cases). Treatment includes dietary and pharmacologic approaches (diazoxide; octreotide) to control hypoglycemia followed by definitive surgical resection (enucleation, subtotal pancreatectomy, Whipple). Refractory/metastatic cases are treated with mTOR inhibitor everolimus. Over 90% of insulinomas are indolent and cured by surgery. About 7-10% are aggressive with tendency to metastasize. Loss of DAXX or ATRX expression is associated with adverse prognosis.