Neurofibromatosis 2
Reviewer(s): Dharam M. Ramnani, MD
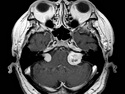
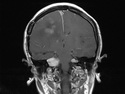
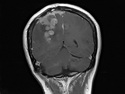
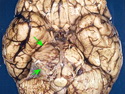
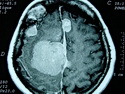
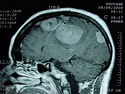
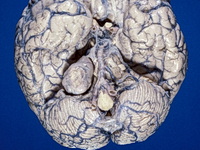
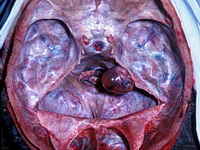
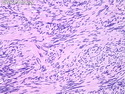
Neurofibromatosis 2 (NF2) is an autosomal dominant disorder affecting 1 in 33,000 to 40,000 individuals. It results from inactivating germline mutations in the tumor suppressor gene NF2 on chromosome 22q12.2. About 50% of NF2 cases have de novo mutations. The type of mutations present (nonsense, missense, frameshift, or large deletions) affect the disease severity. NF2 codes for Merlin (also known as Schwannomin) - a 595 amino acid protein expressed in Schwann cells, meningeal cells, and eye lens. It belongs to moesin-ezrin-radixin family of cytoskeletal-associated proteins. Merlin interacts with several cytoskeletal proteins and cell membrane-associated signaling complexes. It appears to play an important role in cell mobility and contact-dependent inhibition of cellular proliferation. The mechanism by which loss of Merlin function causes tumorigenesis in not exactly understood. The diagnostic hallmark of NF2 is bilateral vestibular schwannomas (acoustic neuromas) affecting vestibular portion of the eighth cranial nerve. They are manifested by tinnitus and hearing loss starting in adolescence or early adulthood. Other manifestations include: cutaneous schwannomas, schwannomas of other nerves intracranially and in the spinal compartment, meningioma, ependymoma, and glioma. There is no risk of malignant transformation in schwannomas of NF2, however they cause significant morbidity and reduced life-span due to their location in brain and spinal cord. Patients develop deafness, vision loss, imbalance and gait abnormalities, muscle weakness, paralysis, pain, and seizures. Reference:
Enzinger & Weiss's Soft Tissue Tumors, Sixth Edition, 2014; p. 829-831. Updated: April 2, 2017.